Quando sospettare la DCP
La caratteristica essenziale dei soggetti da valutare per la diagnosi di DCP è rappresentata dalla loro tendenza al continuo sviluppo di infezioni respiratorie. Questo è da attribuire al ristagno di secrezioni mucose per il deficit del trasporto muco-ciliare proprio della malattia. Infatti, questa condizione si caratterizza per un’alterata o assente motilità delle ciglia di rivestimento dell’epitelio respiratorio che abitualmente assicurano la rimozione delle secrezioni, rappresentando in tal modo il primo meccanismo di difesa contro le aggressioni infettive. La DCP, tuttavia, presenta una notevole variabilità fenotipica e la difficoltà di selezione dei soggetti da sottoporre alle procedure diagnostiche è legata al fatto che non esiste alcuna caratteristica clinica specifica della malattia. Poiché le manifestazioni cliniche della DCP (rinite, tosse, bronchite, otite media) sono comuni alla maggior parte di bambini normali e lo spettro di gravità della malattia è molto ampio, il sospetto diagnostico per la DCP è spesso posto tardivamente. Le espressioni della malattia, inoltre, spesso differiscono per epoca di insorgenza, potendo essere suddivise come più tipiche del periodo neonatale, dell’età pediatrica e dell’adulto. Alcune caratteristiche del feto, come l’eterotassia, devono, tuttavia, indurre a considerare la possibilità della malattia già prima della nascita. Comunque è la tosse catarrale quotidiana e in ogni periodo dell’anno fin dalle prime epoche della vita la manifestazione più comune.
Presentazione clinica nel periodo antenatale
In considerazione della natura genetica della malattia, una storia familiare positiva per DCP o per sintomi respiratori cronico-recidivanti non diagnosticati è un importante elemento per sospettare la diagnosi. Il riscontro durante la gravidanza di eterotassia viscerale, totale (situs inversus) o parziale (situs ambiguus), impone precisi accertamenti e un attento follow-up post-natale, poiché circa un quarto dei soggetti con queste anomalie risulta affetto da DCP. Allo stesso modo, poiché l’idrocefalo neonatale e alcune cardiopatie congenite possono associarsi alla DCP, il riscontro ecografico di una ventricolomegalia fetale e/o di una cardiopatia complessa deve costituire un sospetto clinico meritevole di attenzione.Presentazione clinica nel periodo neonatale
La DCP può manifestarsi alla nascita con distress respiratorio, peraltro inspiegato in un neonato a termine. Alcuni aspetti clinici come l’idrocefalo idiopatico (secondario a disfunzione delle cellule ciliate ependimali), le cardiopatie congenite complesse e l’asplenia/polispnenia possono talora essere associati. Spesso la malattia si rende evidente nei primi giorni di vita con secrezione nasale mucosa o muco-purulenta, tipicamente cronica. Questi quadri neonatali dovrebbero sempre allertare il pediatra sulla possibilità di una DCP e indurlo ad escludere questa condizione.Presentazione clinica nel bambino
I sintomi della DCP mostrano un’espressione clinica variabile in età pediatrica, riflettendo soprattutto l’interessamento dell’apparato respiratorio, e abitualmente tendono a recidivare o a non risolversi. Vi è il coinvolgimento delle prime vie aeree (naso e seni paranasali), ma anche della tuba e dell’orecchio medio. La rinorrea muco-purulenta cronica rappresenta un sintomo molto tipico fin dai primi mesi. Nel corso degli anni il quadro clinico può assumere le caratteristiche della rinosinusite con sintomi più eterogenei, quali rinorrea anteriore e posteriore, ostruzione nasale, febbre, alitosi, cefalea e tosse. Il coinvolgimento dell’orecchio medio è frequente ed è caratterizzato da otiti ricorrenti, versamento endotimpanico persistente e, nei casi evolutivi, riduzione dell’udito. La poliposi nasale può presentarsi già nei primi anni di vita, ma è tipica delle età successive. Il coinvolgimento delle vie aeree inferiori è caratterizzato dalla presenza di tosse cronica, tipicamente catarrale per il ristagno di muco. Il respiro sibilante è raro, ma talora il quadro clinico può presentarsi con prevalenza di tosse e reperto obiettivo toracico di tipo ostruttivo. La presenza di catarro bronchiale si può accompagnare ad atelectasie ricorrenti, infezioni respiratorie e talora polmoniti.Presentazione clinica nell’adolescente e nell’adulto
Nell’adolescente e nell’adulto le manifestazioni cliniche sono identiche a quelle presenti nel bambino, ma abitualmente tendono ad essere di intensità maggiore in quanto riflettono la progressione del danno d’organo. L’interessamento delle prime vie aeree si manifesta con le caratteristiche della rinosinusite cronica. L’alitosi (per infezione cronica della mucosa nasale), l’ipo/anosmia (per alterata funzione dell’area di percezione olfattiva conseguente al danno flogistico della mucosa), l’ostruzione nasale (dovuta alla poliposi) sono comuni. Nell’adolescente e nell’adulto l’otite media è in genere meno frequente che nel bambino, ma la riduzione dell’udito può persistere. L’interessamento del polmone è svelato dalla tosse cronica con espettorato mucopurulento. Le frequenti riacutizzazioni infettive a carico delle vie aeree inferiori contribuiscono infatti al progressivo sviluppo di bronchiectasie, prevalentemente di tipo cilindrico e a distribuzione segmentale, in genere localizzate nel lobo medio, nella lingula e nei segmenti basali. L’infertilità nel maschio (legata alla immobilità degli spermatozoi) e la subfertilità o ripetute gravidanze ectopiche nella femmina (legate all’alterato trasporto dell’ovulo attraverso le tube) rappresentano manifestazioni della DCP esclusive dell’età adulta.Condizioni cliniche associate
Diverse condizioni cliniche possono associarsi a DCP, quali le cardiopatie congenite complesse, in particolare con disordini di lateralità (isomerismo atriale, trasposizione dei grossi vasi, ritorno venoso anomalo, doppia uscita del ventricolo destro, vena cava superiore bilaterale); la polisplenia; il rene policistico; il fegato policistico; l’atresia biliare; la retinite pigmentosa; l’atresia esofagea; l’idrocefalo. Quando tali manifestazioni si accompagnino a sintomi tipici e/o ad una storia familiare significativa devono indurre a sospettare la DCP.Bibliografia essenziale
- Bush A, et al. Primary ciliary dyskinesia: recent advances in epidemiology, diagnosis, management and relationship with the expanding spectrum of ciliopathy. Expert Rev Respir Med. 2012;6:663-82.
- Dehlink E, et al. Clinical phenotype and current diagnostic criteria for primary ciliary dyskinesia. Expert Rev Respir Med. 2016;19:1-13.
- Leigh MW, et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med. 2009;11:473-87.
Elementi clinici indicativi
La diagnosi della DCP poggia innanzitutto sulla presenza di un fenotipo clinico compatibile, anche se le caratteristiche cliniche di tale condizione non differiscono da quelle riscontrate in pazienti con patologia respiratoria cronica di altra origine (per esempio la Fibrosi Cistica e i deficit immunitari), nelle quali le alterazioni ciliari sono secondarie al processo infiammatorio/infettivo. Nei casi in cui è presente un fondato sospetto e siano state escluse le altre cause compatibili con il quadro clinico, alcune indagini di screening possono risultare utili ai fini di un indirizzo diagnostico, anche se la diagnosi di certezza rende necessaria l’analisi delle cellule epiteliali ciliate al fine di valutarne l’ultrastruttura e la funzione presso i centri specialistici, nei quali tali indagini sono disponibili.
In mancanza di una storia familiare o di dati prenatali significativi, la diagnosi deve essere sospettata in presenza di una combinazione di segni e sintomi e tenendo conto dell’epoca della loro comparsa. Elementi clinici indicativi per DCP, ma variabili in rapporto al fenotipo e all’età, sono:
- Distress respiratorio neonatale, soprattutto se dopo alcune ore dal parto, in nati a termine (senza altra causa), con ricovero in Terapia Intensiva Neonatale
- Rinite persistente a esordio neonatale
- Sinusite cronica, soprattutto se interessa tutti i seni paranasali in rapporto all’epoca di pneumatizzazione o si associa all’agenesia o ipoplasia di alcuni di essi
- Situs inversus viscerum o altri disturbi della lateralità
- Cardiopatia congenita, soprattutto con eterotassia
- Idrocefalo
- Tosse catarrale quotidiana anche nel periodo estivo
- Otiti ricorrenti
- Otorrea prolungata dopo inserzione di tubo trans-timpanico
- Bronchiectasie e bronchiolectasie soprattutto se bilaterali e al lobo medio o ai lobi inferiori
- Bronco-ostruzione ricorrente refrattaria al solo trattamento antiasmatico
- Infertilità maschile e subfertilità femminile
Allo scopo di migliorare l’identificazione dei soggetti da avviare alle indagini diagnostiche, è stato validato un questionario denominato PICADAR (PrImary CiliAry DyskinesiA Rule), che si può utilizzare fin dalle prime epoche della vita nei soggetti con tosse catarrale quotidiana insorta precocemente, condizione indispensabile per il sospetto diagnostico di DCP. Somministrando questo questionario il punteggio massimo ottenibile è pari a 14, che rappresenta il risultato della somma dei punteggi relativi ai seguenti items: 1. nascita a termine (2 punti), 2. sintomi respiratori neonatali (2 punti), 3. ricovero in terapia intensiva neonatale (2 punti), 4. difetto della lateralità (4 punti), 5. cardiopatia congenita (2 punti), 6. rinite cronica (1 punto), 7. otiti ricorrenti e/o ipoacusia (1 punto). I pazienti con uno score ≥ 10 hanno una probabilità >90% di avere test diagnostici positivi per DCP, ma è sufficiente raggiungere uno score di 5 perché il questionario abbia una sensibilità del 90% con una specificità del 75%.

Bibliografia essenziale
- Bush A, et al. Primary ciliary dyskinesia: recent advances in epidemiology, diagnosis, management and relationship with the expanding spectrum of ciliopathy. Expert Rev Respir Med. 2012;6:663-82.
- Dehlink E, et al. Clinical phenotype and current diagnostic criteria for primary ciliary dyskinesia. Expert Rev Respir Med. 2016;19:1-13.
- Behan L, et al. PICADAR: a diagnostic predictive tool for primary ciliary dyskinesia. Eur Respir J. 2016;47:1103-12.
Quali sono i test eseguiti presso la nostra struttura
Nei casi in cui è presente un fondato sospetto e siano state escluse altre cause compatibili con il quadro clinico (per es., fibrosi cistica, deficit immunitari), alcune semplici indagini di screening possono risultare utili ai fini di un indirizzo diagnostico. Per la diagnosi di certezza è tuttavia necessario completare gli accertamenti con indagini più sofisticate.
I test di screening
Misurazione dell’ossido nitrico nasale
Vari studi hanno dimostrato che i livelli di ossido nitrico nell’aria espirata ed in particolare quelli misurati mediante campionamento nasale (nNO) sono molto bassi nella DCP. Tuttavia, se il rilievo di livelli normali od elevati di nNO può aiutare ad escludere la DCP, in alcuni pazienti con accertamenti diagnostici positivi i livelli di nNO appaiono normali. Infine, il riscontro di bassi livelli di nNO non è specifico della DCP, ma è presente anche nella fibrosi cistica, nella panbronchiolite, nelle ostruzioni nasali, nella sinusite ed in caso di poliposi nasale. Inoltre, mentre la misurazione del nNO è ben standardizzata nel soggetto collaborante, risulta poco agevole nel non collaborante, per il quale è stato proposto di utilizzare la media dei valori di picco ottenuti in almeno cinque rilievi.Bibliografia essenziale
- Werner C, et al. Diagnosis and management of primary ciliary dyskinesia. Cilia. 2015;4:2.
- Collins SA, et al. The dangers of widespread nitric oxide screening for primary ciliary dyskinesia. Thorax. 2016;71:560-1.
- Shapiro AJ, et al. Accuracy of Nasal Nitric Oxide Measurement as a Diagnostic Test for Primary Ciliary Dyskinesia. A Systematic Review and Meta-analysis. Ann Am Thorac Soc. 2017;14:1184-96.
- Lucas JS, et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J. 2017;49(1).
- Shapiro AJ, et al., American Thoracic Society Assembly on Pediatrics. Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;197:e24-e39.
I test diagnostici
La diagnosi della DCP richiede l’analisi delle cellule ciliate dell’epitelio respiratorio in campioni di mucosa prelevati mediante brushing nasale e si basa sulla valutazione della funzione e dell’ultrastruttura ciliare, mentre nei casi di difficile inquadramento è necessario ricorrere allo studio della ciliogenesi in coltura delle cellule respiratorie ciliate o ai test genetici.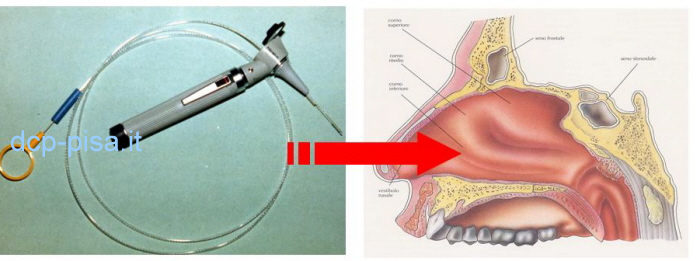
Valutazione della funzione ciliare in vitro o ex vivo
Per eseguire oltre alla valutazione della frequenza del battito ciliare anche un’analisi del pattern motorio, indagini raccomandate per la diagnosi di DCP, è necessario disporre di un sistema di registrazione delle immagini delle ciglia in movimento ad alto ingrandimento con velocità di campionamento sufficientemente elevata da catturare le variazioni reali all’interno del processo. Questo è reso possibile dalla tecnologia che consente l’osservazione dell’evento mediante microscopio dotato di lenti ad immersione ad elevato ingrandimento e la cattura di immagini video digitali ad alta velocità (High-speed Video Microscopy Analysis: HVMA). È necessario eseguire sia la valutazione della frequenza del battito ciliare sia l’analisi del pattern motorio perché nella DCP si possono osservare valori di frequenza del battito ciliare da molto bassi ad estremamente elevati, insieme a varie anomalie del pattern motorio (per esempio movimenti rigidi o circolari) e/o ciglia immobili. Poiché le alterazioni evidenziate nella DCP sono le stesse che si osservano in presenza di un processo infiammatorio, sebbene in misura generalmente inferiore e con il contemporaneo rilievo di più tipi di pattern patologico, è necessario che la valutazione sia quantitativa.


Da Chilvers MA. et al. J Allergy Clin Immunol. 2003; 112: 518-24
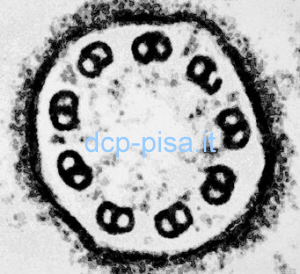
Esame Ultrastrutturale delle ciglia dell’epitelio respiratorio
L’esame ultrastrutturale degli assonemi ciliari mediante microscopia elettronica a trasmissione (TEM) ha un ruolo importante nella diagnosi della DCP, tuttavia è necessario essere consapevoli dei suoi limiti che possono rendere difficile un inquadramento nosologico delle lesioni (che non possono essere correttamente classificate in circa un terzo dei casi). È indispensabile esaminare un numero sufficiente di assonemi adeguatamente sezionati ed appartenenti ad un numero non esiguo di cellule non adiacenti ottenute da diversi frammenti di epitelio intatto, valutando solo gli assonemi sezionati in maniera perfettamente trasversale nella loro parte intermedia e studiando sezioni seriate per essere certi che le lesioni evidenziate non siano focali. L’unico difetto sicuramente congenito, che di solito interessa quasi tutte le ciglia, è il deficit dineinico totale. Infatti, sia l’assenza parziale di entrambi i bracci di dineina sia di isolati deficit del braccio esterno e interno possono anche essere dovuti alla flogosi. Inoltre, il deficit del solo braccio interno è raro ed è molto difficile da diagnosticare a causa dell’intrinseca difficoltà a visualizzarlo essendo sempre meno ben definito rispetto all’esterno. Anche la corretta interpretazione dell’assenza della coppia centrale di microtubuli può risultare particolarmente difficile. Infatti, se questa alterazione può interessare meno di un terzo delle ciglia respiratorie nella DCP, può anche essere presente in più del 30% delle ciglia a seguito di un processo infiammatorio. Altri difetti comunemente considerati di natura congenita, quali la traslocazione, la trasposizione, l’aplasia ciliare e l’alterato orientamento, possono ugualmente essere dovuti ad una flogosi cronica, per cui in questi casi soltanto una valutazione quantitativa delle lesioni (valutazione morfometrica) in un numero elevato di assonemi può orientare verso la diagnosi corretta.
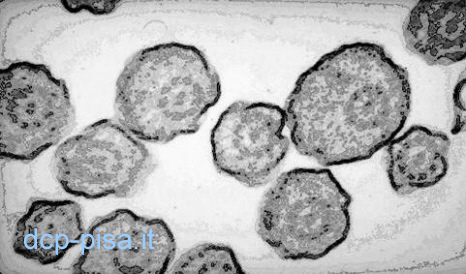
Studio della ciliogenesi in coltura
La valutazione della ciliogenesi in cellule poste in coltura per stabilire la diagnosi di DCP è particolarmente utile nei casi di difficile interpretazione. Infatti, dopo ciliogenesi le cellule epiteliali ciliate ottenute da pazienti con DCP continueranno a presentare un’attività ciliare anormale e conserveranno le stesse alterazioni ultrastrutturali, ma quelle ottenute da pazienti con discinesia ciliare secondaria ad un processo infiammatorio riguadagneranno una funzione ed una ultrastruttura normali. Lo studio della frequenza del battito ciliare e della coordinazione delle ciglia può essere condotto mediante valutazione diretta del movimento delle cellule respiratorie ciliate aggregate in forma di sferoidi nel mezzo di coltura. La DCP può essere esclusa se la polarità e il battito coordinato delle ciglia determina continui movimenti di migrazione o di rotazione degli sferoidi intorno al loro asse. Con tale tecnica gli sferoidi possono essere identificati già dopo 24-48 ore di coltura, ma è opportuno che la valutazione dell’attività ciliare venga protratta fino al 21°giorno quando la formazione di nuove ciglia dovrebbe essere completata.
Bibliografia essenziale
- Lucas JS, et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J. 2017;49(1).
- Shapiro AJ, et al., American Thoracic Society Assembly on Pediatrics. Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;197:e24-e39.
- Pifferi M, et al. Simplified cell culture method for the diagnosis of atypical primary ciliary dyskinesia. Thorax. 2009;64:1077-81.
- Hirst RA, et al. Ciliated air-liquid cultures as an aid to diagnostic testing of primary ciliary dyskinesia. Chest. 2010;138:1441-7.
I test genetici eseguiti presso il nostro ospedale
La DCP è una malattia geneticamente eterogenea che viene trasmessa nella maggior parte dei casi come carattere autosomico recessivo, ma sono noti anche patterns di ereditarietà X-linked. Attualmente circa 50 geni sono stati associati alla malattia, e oltre il 70% dei pazienti testati ha mutazioni bialleliche in uno di questi geni. Questo numero certamente aumenterà con la scoperta di ulteriori geni, giacché si stima che i geni potenzialmente coinvolti nella malattia possano essere anche un migliaio. A molti dei geni mutati oggi noti sono state attribuite specifiche alterazioni dell’ultrastruttura ciliare (geni che codificano proteine del braccio esterno di dineina, del braccio interno di dineina, del complesso regolatorio della dineina, dei ponti radiali e dell’apparato centrale). Più recentemente sono state identificate mutazioni patogenetiche in geni che codificano alcune proteine citoplasmatiche non integrate nell’assonema ciliare, alcune delle quali forma complessi essenziali per il preassemblaggio delle unità motrici della dineina. Le mutazioni in questi geni può, pertanto, essere responsabile di grossi difetti o di sottili alterazioni ultrastrutturali, oltre che di una ultrastruttura apparentemente normale. È anche noto che le mutazioni nei geni che si accompagnano a grossi difetti ultrastrutturali si associano anche ad alterazioni marcate del movimento ciliare e a una randomizzazione dell’asimmetria destra/sinistra degli organi, mentre ciò non avviene quando sono interessati i geni le cui mutazioni si associano ad anomalie ultrastrutturali compatibili con la presenza di movimenti circolari delle ciglia. Inoltre, la retinite pigmentosa X-linked recessiva, i deficit dell’udito di tipo neurosensoriale e la DCP sono state associate a mutazioni nel gene regolatore guanosina trifosfatasi (RPGR) della retinite pigmentosa. Infine, è stata descritta una famiglia con una nuova sindrome causata da mutazioni nel gene della sindrome oro-facio-digitale di tipo 1 (OFD1) e caratterizzata da ritardo mentale X- linked recessivo, macrocefalia e DCP. Le attuali conoscenze delle basi genetiche della malattia non permette, tuttavia, per ora di raccomandare l’indagine genetica come parte dei test diagnostici iniziali, ma suggerisce di eseguirla come test di secondo livello quando la diagnosi di DCP è verosimile, ma non può essere posta dopo aver eseguito l’iter diagnostico già illustrato, oppure come conferma o precisazione diagnostica. Infatti, sebbene sia possibile, con l’avvento delle nuove tecniche di sequenziamento (Next Generation Sequencing), eseguire con maggiore rapidità la ricerca di mutazioni in tutti i geni patogenetici noti, questa, come si diceva, si dimostra in grado di confermare solo il 70% delle diagnosi poste correttamente con le altre procedure, percentuale che scenderebbe drasticamente utilizzando l’indagine genetica sulla base del solo sospetto. Inoltre, le valutazioni genetiche sono ancora costose, poco disponibili e gravate da criticità, quali la necessità di disporre di un’adeguata analisi bioinformatica per filtrare e classificare correttamente le numerose varianti identificate, la difficoltà di interpretazione di varianti con significato incerto, l’impossibilità per la tecnologia di sequenziamento di identificare le mutazioni introniche profonde. Inoltre, le ampie delezioni e le duplicazioni possono essere identificate solo mediante l’impiego dell’ibridazione genomica comparativa su microarray (Array - Comparative Genomic Hybridization o Array-CGH). L’identificazione delle mutazioni nei bambini affetti permetterà di individuare anche lo stato di portatore nei genitori ed in altri membri della famiglia offrendo loro un’opportunità per il consiglio genetico e potrà essere utilizzata come test prenatale nel caso di successive gravidanze. Per la caratterizzazione genetica e per la conferma diagnostica viene eseguita l'analisi mutazionale dei seguenti geni responsabili della DCP:
- CCDC39 (NM_181426)
- CCDC40 (NM_017950)
- RSPH9 (NM_001193341)
- RSPH4A (NM_001010892)
- RSPH3 (NM_031924)
- DNAJB13 (NM_153614)
- DNAH11 (NM_001277115)
- CCDC65 (NM_033124)
- MCIDAS (NM_001190787)
- RSPH1 (NM_080860)
- DRC1 (NM_145038)
- GAS8 (NM_001214)
- CCNO (NM_021147)
- DNAH5 (NM_001369)
- DNAI2 (NM_023036)
- DNAI1 (NM_ 012144)
- NME8 (NM_016616)
- DNAL1 (NM_031427)
- ARMC4 (NM_018076)
- CCDC114 (NM_144577)
- CCDC151 (NM_002743)
- TTC25 (NM_031421)
- LRRC6 (NM_012472)
- DNAAF1 (NM_178452)
- DNAAF2 (NM_018139)
- DNAAF3 (NM_001256714)
- DYX1C1 (NM_130810)
- CCDC103 (NM_001258396)
- ZMYND10 (NM_015896)
- HEARTR2 (NM_017802)
- SPAG1 (NM_172218)
- C21ORF59 (NM_021254)
- PIH1D3 (NM_173494)
- DNAH1 (NM_015512)
- STK36 (NM_015690)
- DNAH8 (NM_0012006927)
- RPGR (NM_00328)
- OFD1 (NM_03611)
- DNAH6 (NM_001370)
- DNAH9 (NM_001372)
- LRRC56 (NM_198075)
- GAS2L2 (NM_139285)
- FOXJ1 (NM_001454)
- C11ORF70 (NM_032930)
- TTC12 (NM_017868)
- CFAP221 (NM_001271049)
- NME5 (NM_003551)
- SPEF2 (NM_024867)
- CFAP57 (NM_001195831)
- NEK10 (NM_199347)
- Horani A, et al. Advances in the Genetics of Primary Ciliary Dyskinesia: Clinical Implications. Chest. 2018 May 22. pii: S0012-3692(18)30754-2.
- Bush A, et al. Primary ciliary dyskinesia: recent advances in epidemiology, diagnosis, management and relationship with the expanding spectrum of ciliopathy. Expert Rev Respir Med. 2012;6:663-82.
- Dehlink E, et al. Clinical phenotype and current diagnostic criteria for primary ciliary dyskinesia. Expert Rev Respir Med. 2016;19:1-13.
- Lucas JS, et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J. 2017;49(1).
- Shapiro AJ, et al., American Thoracic Society Assembly on Pediatrics. Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;197:e24-e39.
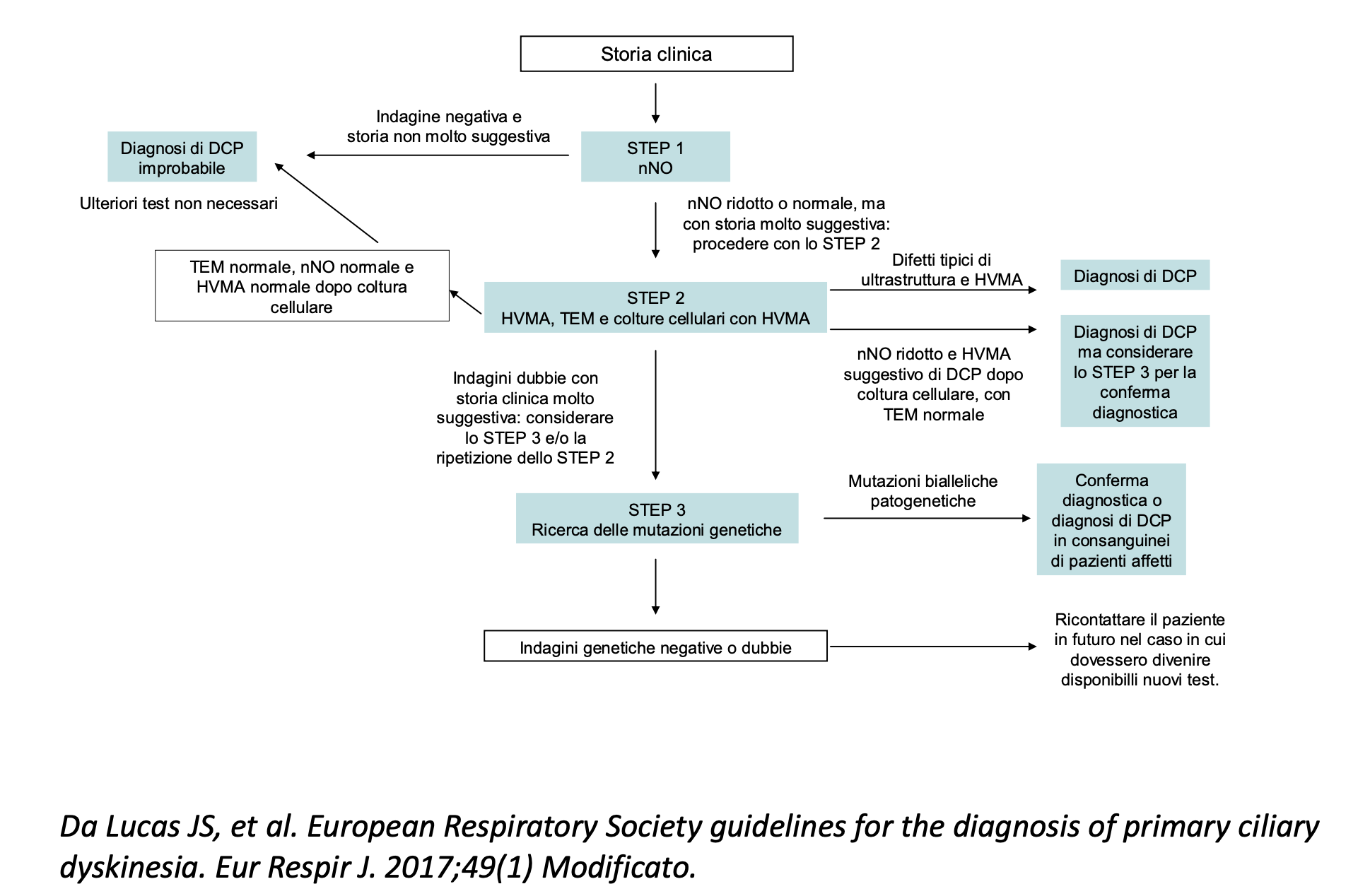
Algoritmo diagnostico utilizzato presso la nostra struttura